






Making decisions about medicines (1)
What is pharmacogenomics?
Pharmacogenomics is the branch of genomics concerned with the way in which an individual's genetic attributes affect their likely response to medicines. When we give a patient a medicine, we want it to have benefit without causing harm. In practice it can be difficult to predict how an individual will respond to any given drug as shown in the image below. Pharmacogenomics can be used to ensure patients receive medicines that are effective while reducing the risk of certain adverse effects.
 |
| Potential patient outcomes when given a medicine Edited image courtesy of HEE Genomics Education Programme |
Pharmacogenomics is important to pharmacy practice as it can help to remove the ‘trial and error’ aspect of treatment choice or effective dosing, ensuring better outcomes for patients. As experts in medicines, the pharmacy workforce are ideally placed to advise on how pharmacogenomic information can be used to adjust or manage a patient’s treatment. An example is hepatitis C (HCV) which has seven distinct genotypes and multiple subtypes. Genotyping of the virus can help when selecting appropriate antiviral therapy.
How pharmacogenomics can influence the response to medicines

• Pharmacodynamics - where a genetic variation reduces binding of the drug to its receptor, thereby decreasing therapeutic efficacy. (e.g. warfarin and VKORC1).
• Adverse reactions – for example altered susceptibility to a hypersensitivity reaction to a certain drug (e.g. HLA-mediated reactions).
• Effects on disease pathogenesis or severity and how they respond to specific therapies (e.g. ovarian cancer and BRCA mutations, NTRK gene fusion in various cancers).
How pharmacogenomics can affect efficacy
1. Pharmacokinetics
Cytochrome p450 (CYP) isoenzymes
In practice one of the most common applications of pharmacogenomics that pharmacy professionals will come across as services develop will involve CYP450 isoenzymes.
Many drugs are metabolised by CYP450 isoenzymes, and there are at least 57 CYPs encoded by the human genome, and these are organised into 18 families, with families 1,2 and 3 responsible for over 50% of the metabolism of common drugs (e.g. CYP2C19, CYP3A4). More information can be found here.
Some medicines are pro-drugs which rely on these enzymes for activation (e.g. clopidogrel), while others rely on these enzymes to break them down and inactivate them (e.g. voriconazole). Individual drugs are not metabolised exclusively by one isoenzyme, although one often predominates.
Many drug – drug interactions are mediated by these enzymes (see the Interactions tutorial).
CYP450 isoenzymes are subject to genetic polymorphism which can result in changes in the enzyme activity. There can be many variations in a specific isoenzyme, some having no appreciable effect, whereas others can have a pronounced effect on how an individual patient metabolises a particular drug. The expression and activity of CYPs can vary considerably among individuals and ethnicities.
This can lead to instances of loss of efficacy or decreased plasma levels, increased plasma levels or adverse effects, some of which may require dose changes or a need for alternative therapies depending on the impact.
Alleles and diplotypes
Most reporting of genetic polymorphism of CYP450 isoenzymes is done in terms of alleles and diplotypes and uses the star (*)-allele nomenclature.
Each allele is defined by a genotype at one or more specific single-nucleotide polymorphisms resulting in variable enzyme activity. Each different variant has a different *number and, whilst nomenclature can vary, these are usually divided into increased activity, active, decreased activity, or loss-of-function variants. *1 is used to denote the ‘wild type’ variant, which would represent ‘normal’ function.
The combination of the same alleles from each chromosome pair is used to determine a patient's diplotype (sometimes also referred to as genotype), which can be used to infer an individual’s metaboliser status. A diplotype of *1/*1 is usually inferred to be a ‘normal’ status.
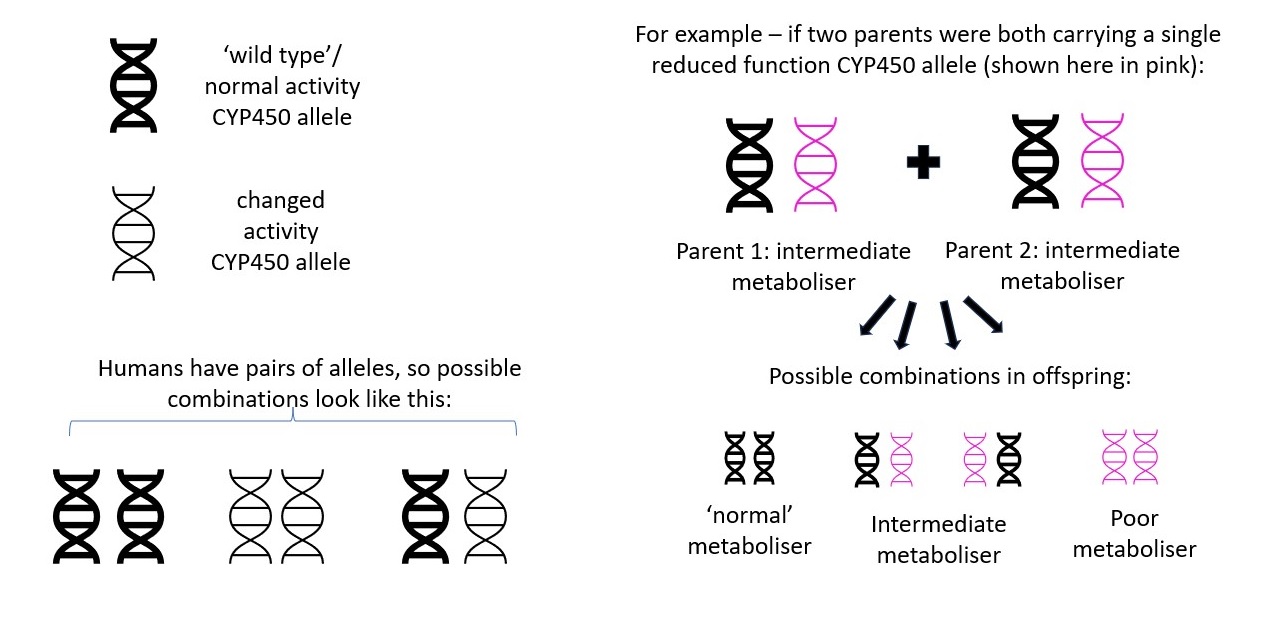 |
| How metaboliser status is passed from parent to offspring Image courtesy of Dr Hayley Wickens, Consultant Pharmacist Genomic Medicine |
Different combinations of alleles can result in the same metaboliser status so in practice diplotypes are usually assigned one of the following functions to describe a patient’s metaboliser status for that specific CYP450 isoenzyme:
• Poor metaboliser
• Intermediate metaboliser
• Normal metaboliser
• Extensive / rapid metaboliser
• Ultra-rapid metaboliser
The reliability of the predicted genotype ‘normal metaboliser’ will depend on the number of variants tested for. The more variants analysed the stronger the prediction as rarer variants are more likely to be picked up. • Intermediate metaboliser
• Normal metaboliser
• Extensive / rapid metaboliser
• Ultra-rapid metaboliser
A caveat is that around 80% of data in genome-wide association studies are based on individuals of European ancestry. Some CYP variants occur at different rates in different ethnic groups. This means that tests that only identify the most common variants in European populations may not be as accurate for non-European populations. Efforts are being made to increase the proportion of genomic research and clinical data coming from patients of non-European ancestry. More information on this can be found here.
CYP2D6 is an example isoenzyme with hundreds of known genetic variants. Codeine is a pro-drug, which relies on metabolism by CYP2D6 to its active metabolite morphine for its analgesic effect. Poor metabolisers of CYP2D6 -> convert less codeine to its active metabolite - > reduced analgesic effect -> poor pain control.
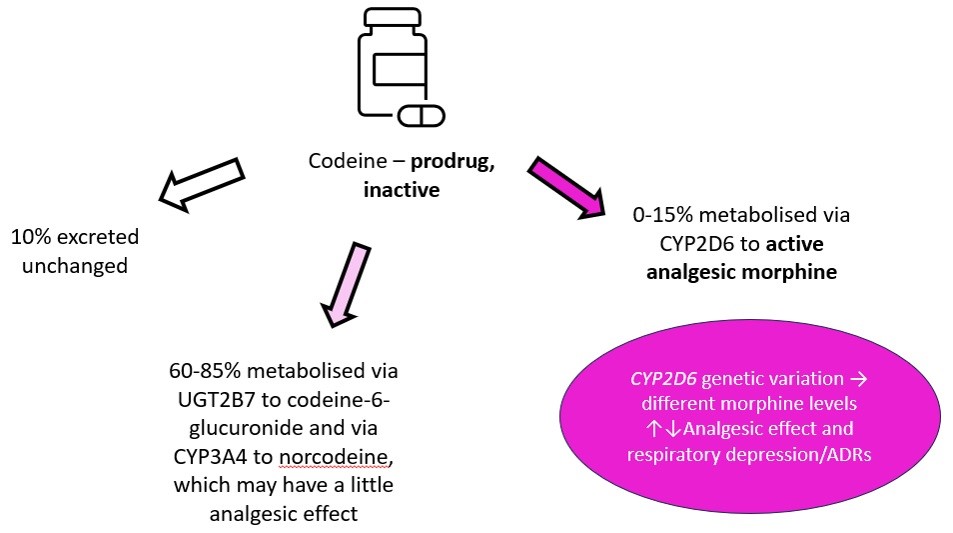 |
| Metabolism of codeine Image courtesy of Dr Hayley Wickens, Consultant Pharmacist Genomic Medicine |
2. Pharmacodynamics
Genetic variation can lead to differences in therapeutic response even when the level of drug itself is unchanged.
These pharmacodynamic effects are often more complex than pharmacokinetic effects. This is in part because of the complexity of drug target pathways compared to pharmacokinetic ones.
In practice this means that for now there are fewer practical clinical applications based on genetic variations in drug pharmacodynamics.
VKORC1 encodes the vitamin K epoxide reductase protein. This enzyme catalyses the conversion of vitamin K-epoxide to vitamin K. Warfarin acts as an inhibitor of VKORC1 reducing the amount of available vitamin K for clotting proteins.
Variants in this gene can alter a patient's sensitivity to warfarin, with common variants in VKORC1 accounting for up to 30% of stable warfarin dose variance in people of European ancestry. Different variants have been found in populations of different ancestry. Genomic effects on warfarin are multifactorial, but in combination with specific CYP2C9 diplotypes, VKORC1 status has been used to guide optimal initial warfarin dosing. However due to population differences these data cannot be assumed to apply across the board. Testing isn’t routinely offered in the UK.
Variants in this gene can alter a patient's sensitivity to warfarin, with common variants in VKORC1 accounting for up to 30% of stable warfarin dose variance in people of European ancestry. Different variants have been found in populations of different ancestry. Genomic effects on warfarin are multifactorial, but in combination with specific CYP2C9 diplotypes, VKORC1 status has been used to guide optimal initial warfarin dosing. However due to population differences these data cannot be assumed to apply across the board. Testing isn’t routinely offered in the UK.
3. Cancer care targeted therapy and personalised medicine
Personalised medicine or targeted treatment is becoming more common in cancer care. Genetic variation of cancer cells can be used to select a more targeted therapy. Some variants can make a person more, or less, likely to respond to a specific treatment.
For example, variants in the epidermal growth factor receptor (EGFR) gene can change how a tumour will respond to EGFR-inhibitor drugs.
In lung cancer, treatment may differ depending on if there are changes in the EGFR gene or the anaplastic lymphoma kinase (ALK) gene. Melanoma treatment can differ depending on whether there are changes in the BRAF gene.
Chronic myeloid leukaemia (CML) is usually characterised by an abnormal BCR-ABL fusion gene on chromosome 22 (Philadelphia chromosome). An abnormal BCR-ABL results in an ’always on’ tyrosine kinase enzyme with associated effects on cell proliferation.
In CML tyrosine kinase inhibitors (TKI) specifically target the ATP-binding site of BRC-ABL, reducing progression of chronic disease to the acute phase and improving survival. Imatinib was the first TKI of this kind, but this was followed by second generation drugs such as nilotinib and bosutinib. However resistance to some TKIs can develop through the T315I gene mutation. Knowing if this mutation is present or not, can help guide treatment choice.
In CML tyrosine kinase inhibitors (TKI) specifically target the ATP-binding site of BRC-ABL, reducing progression of chronic disease to the acute phase and improving survival. Imatinib was the first TKI of this kind, but this was followed by second generation drugs such as nilotinib and bosutinib. However resistance to some TKIs can develop through the T315I gene mutation. Knowing if this mutation is present or not, can help guide treatment choice.